



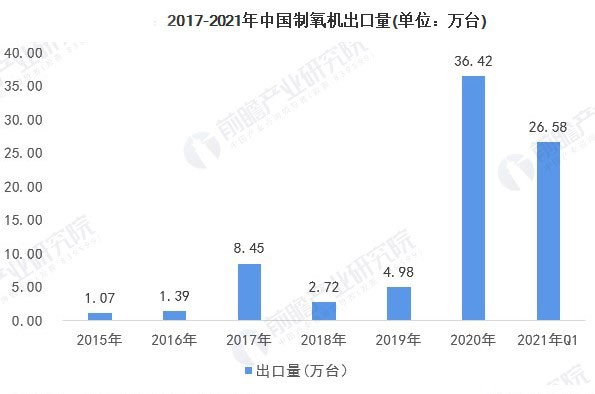
印度疫情失控,制氧机出口暴增 医疗器械出口全球市场准入条件
近年来,我国医疗器械企业国际化步伐加快,在巩固欧美、日本等传统对外贸易市场的基础上,开发新兴市场的意愿不断加强。我国已经成为医疗器械制造大国和出口强国,医疗器械产品出口200多个国家和地区,亚洲、欧洲和北美洲是重点出口区域,拉丁美洲、“一带一路”沿线国家和地区、金砖国家等成为我国医疗器械企业瞄准的新兴市场。
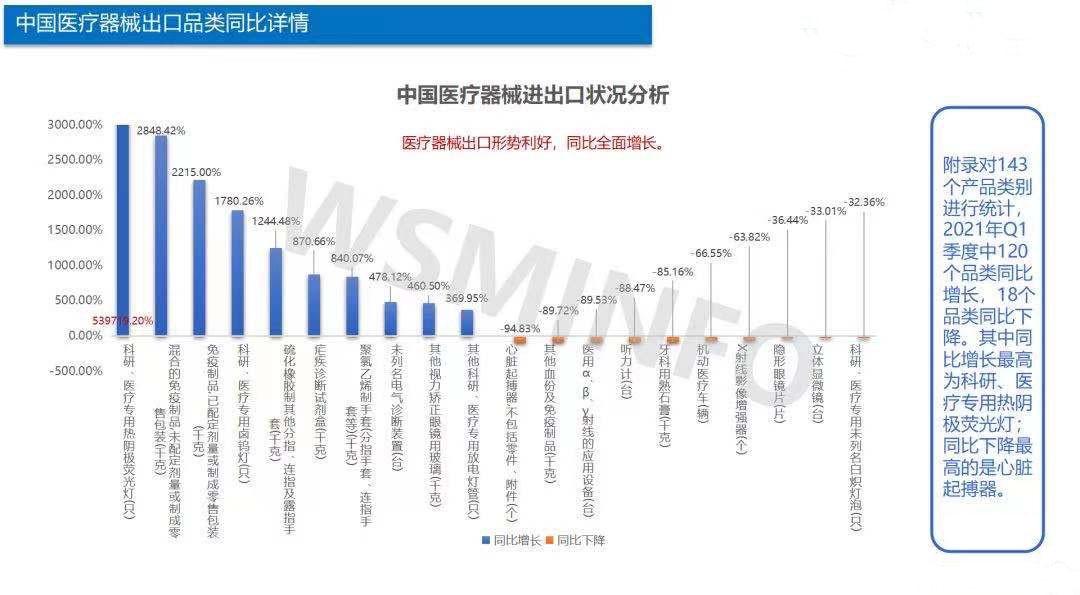
2021年Q1中国进出口总市场容量约437.78亿美元,2021年进出口总额较2020年同比增长76.64%
-2021年Q1中国出口医疗器械约298.09亿美元, 同比2020年出口总金额增长125%
-2021年Q1中国进口医疗器械约139.69亿美元, 同比2020年进口总金额增长21.10%
2020年Q1受疫情影响较大,4月开始复苏,5月达到全年出口峰值。2021年Q1波动频率平稳,未来疫情物资出口情况待观察。
数据来源:中国海关、维斯马
数据来源:中国海关,前瞻经济产业研究院
2021年6月期起又有多项外贸新规落地实施,包括我国发布出口管制新规、新版《医疗器械监督管理条例》实施、印度取消医用氧气等18项产品进口关税、巴基斯坦免除进口氧气及相关设备关税、巴西延长防疫物资进口免税期、欧盟强制实施新版医疗器械法规、欧盟发布木质包装材料最新进口要求、欧亚经济联盟对光伏材料进口实施零关税等。
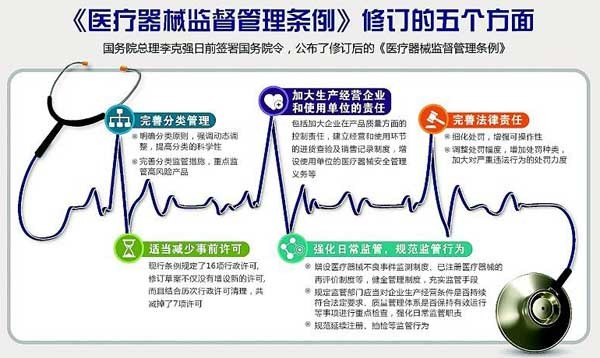
《医疗器械监督管理条例》2021新版
《医疗器械监督管理条例》阐述了医疗器械的定义、目的,分类管理以及备案和注册。
——
第一百零三条:
美国医疗器械产品的市场准入
医疗器械进入美国市场的途径分为:豁免;510(k);PMA。
欧盟医疗器械产品的市场准入
欧盟医疗器械法规提出了“经济运营商”的概念,经济运营商是指制造商、授权代表、进口商、经销商以及任何对系统或手术包类器械进行组合或消毒并投放市场的自然人或法人。即在符合法规规定情况下负责器械生 产(包括组合或灭菌)、销售及上市后运营的自然人或法人。
澳大利亚医疗器械产品的市场准入
TGA 管辖的医疗用品注册处将医疗器械分为三类进行管理,分别为豁免、备案和注册。无论哪类医疗器械,其上市销售前必须得到澳大利亚政府的 准许,符合医疗器械的基本要求,按照符合性审查程序进行审查。对于具有高风险的医疗器械,其质量、安全性、有效性需由TGA评估并在上市前 批准,批准后作为注册产品进入医疗用品注册系统,对其进行编号管理。而一些低风险的医疗器械,由企业自行进行评估,只要符合质量和安全条 件即可进入市场,但要提供相关文件证明其安全有效,并进入医疗用品注册系统,进行编号管理。大多数的器械按备案方式进行管理,通过简要评 估检查是否符合生产、标签以及质量标准。
加拿大医疗器械产品的市场准入
加拿大医疗器械评估体系称为 CMDCAS。加拿大卫生署认定的有资格的 认证机构(即基于 CMDCAS 的认证机构)能提供质量体系认证证书。加拿大 医疗器械法规规定,Ⅱ类、Ⅲ类、Ⅳ 类医疗器械都必须具有质量体系认证 证书。自 2019 年 1 月 1 日起,将以“医疗器械单一审核程序(MDSAP)”强 制全面替代 CMDCAS 成为医疗器械准入的审核程序。未获得 MDSAP 认证证书 的产品将不能在加拿大销售。
日本医疗器械产品的市场准入
按照日本《药事法》规定,一家生产厂的每一种产品,都必须取得厚生 省的生产或入市批准。此外,生产厂还须取得地方政府的生产或入市许可。按修改后《药事法》规定,不仅产品的生产要经厚生省批准,产品的入市 也要经厚生省批准。对任何一种产品,都要求公司须获得生产批准和入市 许可。
巴西医疗器械产品的市场准入
按照巴西政府的规定,经营任何涉及人体的产品(如药品、医疗器械、 美容化妆品等),出口商必须事先向巴西卫生部提出书面申请,且必须通过官方指定的注册持证人提供注册材料。注册材料必须采用葡萄牙语。
俄罗斯医疗器械产品的市场准入
俄罗斯要求医疗器械产品必须进行注册,同时产品还必须通过俄罗斯国 家标准认证后才能上市销售和使用。
印度医疗器械产品的市场准入
根据《药品和化妆品法》规定,对以下十类产品必须按规定的程序进行注册和进口许可:1)心脏支架,2)含药支架,3)导管,4)心脏瓣膜,5) 角膜镜,6)注射器,7)静脉输液针,8)骨粘合剂,9)整形外科植入物, 10)人工假体。2009 年 3 月 CDSCO 又将无菌医疗器械纳入上市注册产品名单,如脊椎针头、胰岛素注射器、器官插管、心脏补片等。
新加坡医疗器械产品的市场准入
《健康产品法令》自 2007 年开始生效后,新加坡对医疗器械的监管力度不断加强,并分阶段实施相应的监管策略。2010 年起对 C 级和 D 级的医疗器械采用强制性审批的监管方式,2012 年起将强制性审批的监管方式扩 大至 B 级和 A 级的医疗器械产品。除了豁免产品,所有类别的医疗器械产 品都必须经过 HAS 注册方可在新加坡上市销售。
马来西亚医疗器械产品的市场准入
对医疗器械产品的上市采取注册制,但对获得美国 FDA 颁发的医疗器械 证书或欧盟 CE 证书的产品的质量是认可的。
菲律宾医疗器械产品的市场准入
菲律宾通过医疗器械企业的营业执照和注册许可的方式进行管理。菲律宾进口所有的医疗器械,进口器械无需事先批准,可以通过地方代理商来 办理相关的注册程序。代理商需要在 BFDA 登记。














